dgame wrote:
正式二 三期,不是比較劣化的實驗結果...高端送的只有跟AZ 比較的非劣化實驗結果吧
不必對比AZ的數據 ,在高端第一期就已經做了
以當前施打新冠疫苗的緊迫性為由,要先走完三期試驗再推出疫苗會追不上變種病毒的腳步,況且三期試驗也仍然在進行當中。
反而, 對比通過EUL的科興? 以及國藥是在實驗於第二期時, 就開始大量施打 ? 這樣沒問題 ?
況且在科興疫苗三期完成後 , 保護力被證明只有51% ? 這樣也沒問題 ? 反正通過了EUL?
正式二 三期,不是比較劣化的實驗結果 ?
啥?比較劣化的實驗結果 ?
所謂的 非劣性試驗(non-inferiority trials), 根本就不是某些媒體, 自行解讀字面的意思, 它的目的就在於檢定新藥療效至少不輸舊藥。
新藥非劣於舊藥的意思
dgame wrote:
上面有給你BBC 當初的報導 跟我記憶中一樣 是使用免疫橋接方式完成"二期"
應該反過來問BBC , 為什麼不能 "免疫橋接" ?
這樣舉例 "免疫橋接"好了
有「台灣抗瘧之父」之稱的連日清老師 , 他不是醫學系畢業的, 只是師大英語系畢業,但是他能在國立臺灣大學公共衛生學院環境衛生研究所兼任教授,在醫學院教病媒蚊, 是很多醫生的老師, 為什麼 ?
因為他的專業, 瘧疾與登革熱的研究, 可比擬正式醫學院畢業的老師
他的教學成就 , 能對比 正式醫學院畢業的老師
再說 恩里科·費米Enrico Fermi 被譽為「原子能之父」。他擁有數項核能相關專利,並在1938年因研究由中子轟擊產生的感生放射以及發現超鈾元素而獲得了諾貝爾物理學獎。
論學歷?論考試資格? 他連博士學位都沒有, 他只是義大利比薩高等師範學校畢業
但是, 費米能不能比擬博士 ? 他的實驗室可是培養了許多博士,諾貝爾獎得主。門下博士生包括歐文·張伯倫、傑弗里·丘、傑爾姆·傅利曼、馬爾溫·戈爾德貝格爾、李政道、阿瑟·羅森菲爾德以及薩姆·特雷曼等人。
對比 , 比較費米Enrico Fermi 的科學成就, 絕對超越許多大學的博士
不知道您對 "中和抗體" 有沒有概念 , 無論是 多株抗體 或單株抗體 , 抗體能夠先和病毒S蛋白結合 , 病毒就無法嵌合上人體細胞 。
這樣的抗體稱為中和抗體 , 是有效的抗體
雖然抗體有效 , 但若抗體的數量不足 , 只中和了某些病毒 , 仍然存在許多未被抗體中和的"空檔"病毒 , 這些無"抗體"防守的病毒就會造成感染
抗體 力價 必須高於 病毒的毒力 , 才有保護力
關於抗體"效價" ,抗體力價夠不夠力? ,是使用 "滴定度" 來評估 ,"滴定度"反映的是抗體結合抗原特異性位點的最低濃度。
方法是 不斷稀釋血清,直到最後抗體和血清樣本不再呈現陽性,這個稀釋多少倍數的"最低濃度"即滴定度,被用來表示抗體的數量是否足夠 , 能否中和病毒。中和病毒 就有保護力
過去的"流感疫苗"也常用橋接方法來監視,明年要用那一株流感?、有什麼樣的病毒變異?,抗體中和"滴度"數等,都拿來對比, 用這樣的模式去看下一年可能需要的疫苗種類
依據橋接所公布的數字中,科興疫苗中和抗體倍數為0.2倍,再來是AZ的0.5倍,而高端呈現的資料,與抗體中和數字是1.7倍,和AZ的0.5倍計算下來就是3.4倍,「保護力只比輝瑞跟莫德納疫苗稍微低一點」。
南韓SK新冠疫苗也將與AZ疫苗進行比對性試驗,採免疫橋接方式
前面已經說明 免疫系統 的特性 , 累積了"免疫記憶 " , 下一次的反應就能啟動速度更快,反應強度更大
免疫系統 需要 "補強" ,
這樣舉例好了 , 免疫系統如同身體的警察
通緝犯(病毒)的資料(疫苗抗原或來自舊冠毒的類似抗原)如果沒送到戶籍地 ,警察(免疫系統)不會去抓他(病毒)
通緝犯的資料送進來 , 如果沒有照片 , 警察不知道通緝犯的長像 ,警察想抓也不知如何去抓他?
如果只送一次 通緝犯(病毒)的資料 , 戶籍地警察(免疫系統) 記不住 , 補強注射第二劑 ,第三劑 , 就是再送一次 通緝犯(病毒)的資料 , 讓戶籍地警察(免疫系統) 補強記憶能記住。
這樣 戶籍地警察(免疫系統)記住了通緝犯(病毒), 才能展開追捕
以當時搶不到疫苗的情況 , 高端疫苗至少多了一種選擇 , 多一種選擇而不是強迫注射, 補足了缺口 ,而且有了自己製造「蛋白質次單位疫苗」的經驗
這樣和美國國衛院合作的經驗很寶貴, 如同我國自製的 IDF 經國號戰機 , IDF 經國號戰機 也只有台灣在使用, 此「自製防禦戰機」是比不上 F16 , 但是在當時補足了F5E老舊的空防缺口, 且 IDF 的出現才促使美國同意出售F16 。
以現在事後來看 , 國外都稱許 台灣的疫情控制很好 , 離中國這麼近 ,卻沒有大規模封城 , 經濟活動都還能進行,是疫情影響最小的國家 。
再者,只看第幾期? 卻不看實驗內容到底做了什麼?
如同只看 費米Enrico Fermi 是比薩高等師範學校畢業 , 連日清老師是師大英語系畢業
對於生物製劑如何審核上市? 以及如何EUA緊急授權 的流程 , 都存在"不明所以"的誤解
事實是 , EUA 的要求已經做到傳統第三期的內容了
(EUA) 緊急授權不同於以往商業上的申請程序,美國的EUA 降低了原本的標準 無論在第一期→第二期 →第三期 , 時程,人數 , 測試項目 ,都是大幅縮水的
例如,輝瑞通過緊急授權是2020年的12月12日 , 所謂的第三期當時只完成藥廠的內部初步試驗 , 正式的第三期報告根本沒有完成 , 這樣的報告是無法通過FDA的正式審查
是由FDA 專家小組背書 而取得緊急使用授權 ,其疫苗有效性及安全性符合緊急使用授權 (EUA) 申請, 已經降低了的標準。但是在時程上是提早 。
通過緊急授權時 , 輝瑞第三期試驗同步仍然在進行當中
又例如2020年12月18日,Moderna mRNA-1273獲得FDA核發緊急使用授權, 這並非臨床試驗的最終結果,因為莫德納的臨床試驗計劃是要到2022年末才會有正式結論。
EUA的流程,不同就於傳統的一期人體試驗,第一期只需50到100人,主要進行疫苗的安定性探索,二期則是,約200人到500人,進行疫苗的劑量探索,到了三期,才會進行大規模測試。
EUA在一期的時候,已經在進行傳統二期的測試內容,二期的人數也來到3500人以上,也就是說,二期已經在進行傳統三期的大規模測試。
以美國FDA的標準 , 申請EUA提交的文件包括該疫苗進行的第1階段和第2階段研究積累的所有安全性數據,並希望第3階段數據包括至少平均有 2個月的追蹤follow-up(這意味著在完成完整的疫苗接種方案後,至少有一半的3期臨床試驗疫苗接受者有2個月的追蹤查訪。)
美國 FDA 對於第三期 數據的標準是 超過 3,000 名疫苗注射受測者
高端疫苗在進行二期臨床試驗,收案人數超過原預期的 3700 人,這就已經達到 美國FDA 要求的第三期 最低標準 3000人 , 總共收案 4000 多人,超過衛福部建議取得台灣緊急使用授權 (EUA) 受試者人數要求,且4月底共 3700 名受試者已完成第二劑接種。
這樣 5 月底將有臨床試驗分析結果,6月就 2個月的 follow-up 了,依法規申請 EUA
高端 採用的就是 FDA 要求的第三期條件去做 第二期
https://www.fda.gov/media/142749/download
Emergency Use Authorization for Vaccines to Prevent COVID-19
Guidance for Industry
Document issued on May 25, 2021.
這是 FDA 準則 建議的 第三期標準

以美國FDA的標準 , 申請EUA提交的文件包括該疫苗進行的第1階段和第2階段研究積累的所有安全性數據,並希望第3階段數據包括至少平均有 2個月的追蹤follow-up(這意味著在完成完整的疫苗接種方案後,至少有一半的3期臨床試驗疫苗接受者有2個月的追蹤查訪。)
美國 FDA 對於第三期 數據的標準是 超過 3,000 名疫苗注射受測者
FDA預期的 EUA要求將包括一個超過3,000名疫苗接種者的3期安全性數據庫,代表參加該3期研究的參與者中有很大比例的人受到了嚴重不良事件和特別關注的不良事件的關注在完成完整的疫苗接種方案後至少一個月。
國產疫苗第二期就是依據 這個第三期標準 去 設計的
高端在4月底共 3700 名受試者已完成第二劑接種, → 6月就 2個月的 follow-up 了
這第二期就已經達到 美國FDA 要求的第三期 最低標準 3000人
4月底共 3700 名受試者已完成第二劑接種
4月 → 6月底 , 有近兩個月的時間 , 美國FDA 要求的第三期為何無法申請EUA ????
不看受測內容為何 ? 卻只看冠上標籤第三期?
誰承認?誰不承認這些主觀認定? 卻不去看內容為何 ? 實質的實驗項目是什麼 ?
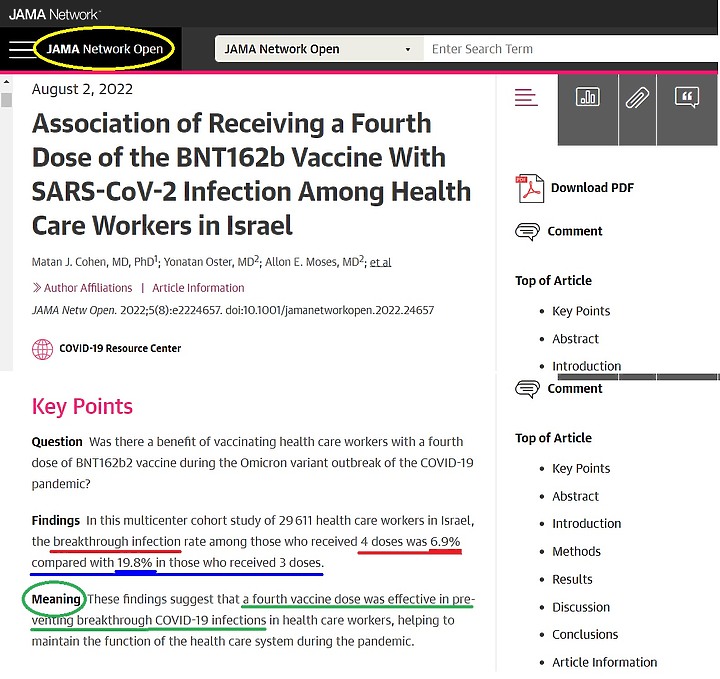
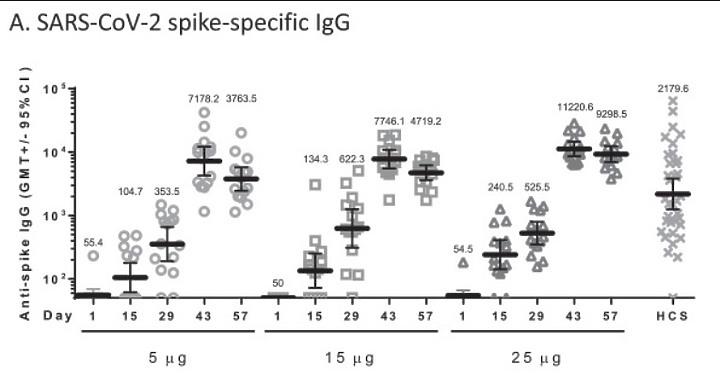
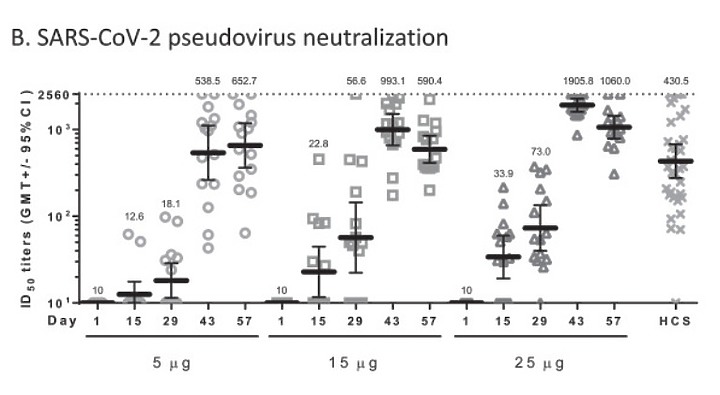
是否應該回到實質內容, 客觀的科學數字來看高端疫苗的 Nab INF--γ ,IL-4 為多少?
於 95% 置信區間的最低幾何平均 (GMT) 比率應至少為 0.67 , 實驗算出來, 經專家會議認定
去年2021有二篇報告
將自然感染復原後的血清, 和不同種類的疫苗注射後所產生之中和抗體 , 比較後做成線性關係圖
第一篇

以 convalescent 自然感染恢復期抗體 為標準 ,neutralisation titre設為 1
二篇不約而同有類似結果
中和抗體效價最差的就是inactivated不活化疫苗 CoronaVac , 中間的就是 Ad-vectored腺病毒載體 , 再高一點是subunit 次單位蛋白疫苗,最高是m-RNA疫苗

在Y軸 50%中和抗體 對比疫苗效力(最低標)就落在X軸約0.67
WHO尋求 COVAX 第三期試驗的標準時
COVAX 由流行病預防創新聯盟 (CEPI)、全球疫苗免疫聯盟 (Gavi) 和世界衛生組織 (WHO) 以及主要交付合作夥伴聯合國兒童基金會共同領導。其目的是加快 COVID-19 疫苗的開發和生產,並確保世界上每個國家都能公平公正地獲得疫苗。
CEPI 就有這樣的要求最低標準

高端GMT是高於達 3.4 , 在 Subunit (次單位疫苗)那個框框的位置 , 效力約 90

 對岸好歹有過耶
對岸好歹有過耶
 二期數據優異三期翻車的比爾蓋茲投資的那一支夠出名了吧
二期數據優異三期翻車的比爾蓋茲投資的那一支夠出名了吧 科興都差點過不了.拿別人不認的流程來跑,不會顯示比較高明
科興都差點過不了.拿別人不認的流程來跑,不會顯示比較高明